Tyler Ford
12/04/2019
In previous blog posts, we’ve discussed the importance of expanding the CRISPR toolkit. TLDR: different CRISPR systems are best suited for different applications.
This is all well and good, but you might be wondering – How do researchers expand the CRISPR toolkit?
In this post, we’ll discuss how researchers analyze bacterial and archaeal genomes to find new CRISPR systems.
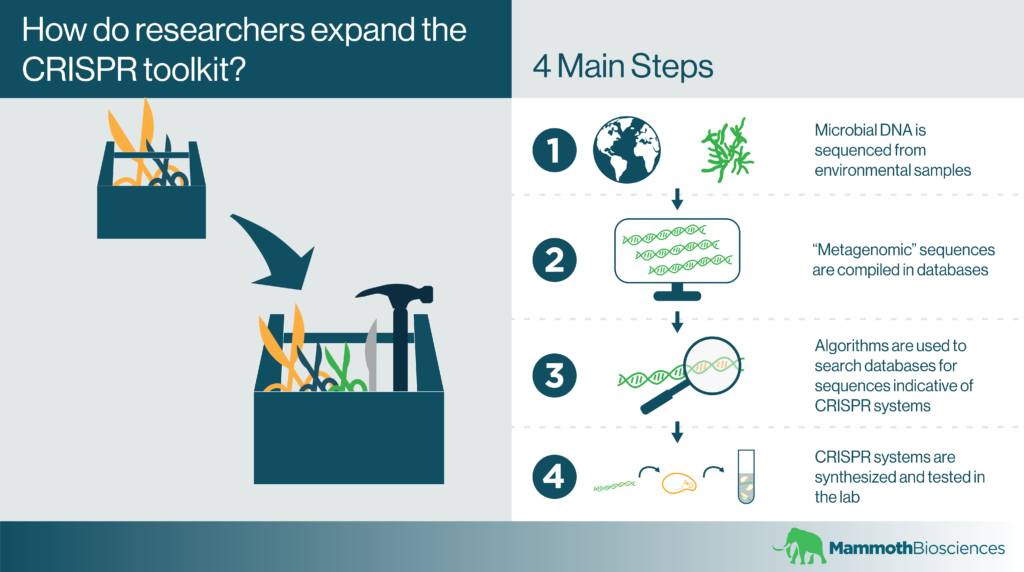
Catalyzing the search process through similarities between CRISPR systems
All functional CRISPR systems come with similar components:
- Cas “effector” proteins that cut nucleic acid sequences as directed by RNA guides (think Cas12a, Cas13, and Cas14)
- CRISPR arrays – these repeating stretches of DNA encode the guides that tell Cas effector proteins where to cut
- Proteins that insert new guide sequences into CRISPR arrays – the most common protein involved in this process is Cas1
Although they ultimately carry out similar functions, the Cas effector proteins from different CRISPR systems can be highly dissimilar in terms of size and composition. As a result, it’s hard to identify new CRISPR systems based on Cas effector proteins.
CRISPR arrays and Cas1 are much easier to identify across CRISPR systems. Thus, to find new CRISPR systems, researchers often start by looking for bacterial DNA sequences encoding CRISPR arrays or Cas1. Then, they check to see if any nearby DNA sequences encode CRISPR effectors.
Finding new CRISPR systems in uncultured bacteria through metagenomics
CRISPR systems are found in many bacteria and archaea. Yet, researchers have only cultured a small fraction of natural bacterial and archaeal strains. At first glance, you might think this would make it very difficult to mine microbial genomes for new CRISPR systems. Luckily, researchers don’t actually need to grow these microbes to find and characterize their CRISPR systems.
Genome sequencing technologies have advanced far enough that it’s possible to sequence microbial genomes directly from environmental samples. After these so-called “metagenomes” are compiled, scientists generally deposit their sequences in databases. Databases like the NCBI’s Microbial Genomes Database contain genomes isolated from a wide range of interesting habitats. These range from hot springs, to camel rumen, to corals, and beyond!
After accessing these databases, researchers can develop algorithms that search the deposited metagenomes for the hallmarks of CRISPR systems (CRISPR arrays and Cas1). Using such automated search processes, they can find new CRISPR systems without culturing the microbes they come from.
As an example, the search process could look something like this:
- Search metagenomic data for DNA sequences encoding CRISPR arrays or Cas1
- Check for CRISPR effectors near the identified sequences
- Synthesize promising CRISPR effector proteins and express them in suitable lab strains
- Test these proteins for functionality in vitro
- Further characterize the new effectors as necessary
Once researchers find a promising Cas effector (i.e. one that actually cuts DNA in vitro or in a lab strain), they can use its sequence to seed a second metagenomic search. This enables them to find homologous systems; these might encode effector proteins and guides that are easier to express, that target different nucleic acids, or that come in different sizes.
Working to expand the CRISPR toolkit for diverse applications
Of course, things get a little more complicated than this when it comes to doing the actual computational and lab work. Nonetheless, the Mammoth Team is excited to use similar techniques to expand the CRISPR toolkit. We look forward to partnering with other researchers and organizations to develop these new systems and their applications!
